
High-Throughput DNA Sequencing To Profile Microbial Water Quality Of Potable Reuse
By Menu B. Leddy, Megan H. Plumlee, Rose S. Kantor, Kara L. Nelson, Scott E. Miller, Lauren C. Kennedy, Blake W. Stamps, John R. Spear, Nur A. Hasan, Rita R. Colwell
Research scientists are making strides to advance the safety and application of potable water reuse through the use of metagenomics for water quality analysis.
Due to recent advances in high-throughput sequencing methods, identifying complete microbial communities present in environmental samples has become feasible, including those in water and biofilms. High-throughput sequencing refers to methods that sequence deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) at an unprecedented speed, with greater coverage (amount sequenced), and at a lower cost than previously possible. Also called next-generation sequencing, high-throughput sequencing is being used for the first time to evaluate the microbial water quality of purified recycled water, further advancing the practice of potable reuse and providing fascinating insights not possible with previous methods.
Potable Reuse And Microbial Water Quality
Many communities in the U.S. and around the world practice water reuse. Water is used more than once for beneficial purposes including drinking (i.e., potable use), irrigation, and industry. Wastewater is a valuable resource that can be appropriately treated and (re)used for potable and nonpotable applications in many regions where water demand exceeds supply. Potable reuse has gained significant attention and acceptance, and it is made possible through a combination of advanced water treatment technologies that together produce highly purified finished water. Technologies include ozone, biological activated carbon, microfiltration (MF), reverse osmosis (RO), and ultravioletbased advanced oxidation processes (UV-AOP), of which a subset or all can be employed in an engineered treatment train to produce high-quality water.
In potable reuse, purified recycled water can be blended with the source water delivered to a conventional drinking water treatment facility or blended with finished drinking water. One form of potable reuse is groundwater augmentation, in which reclaimed water is percolated or injected into a groundwater aquifer to supplement a groundwater-based drinking water supply. One example is the Groundwater Replenishment System (GWRS) operated by Orange County Water District (OCWD) in Fountain Valley, CA. OCWD purifies local municipal wastewater, using advanced treatment to generate high-quality water that meets drinking water standards, and then injects and percolates the water into the ground. At downgradient locations, groundwater is pumped from the aquifer via production wells as a drinking water supply and then conventionally treated prior to use (e.g., disinfection). Some communities are evaluating more direct schemes in which purified recycled water is introduced into a drinking water treatment plant at the raw water supply (i.e., no groundwater storage) or blended with the treated water from the plant (e.g., El Paso, TX).
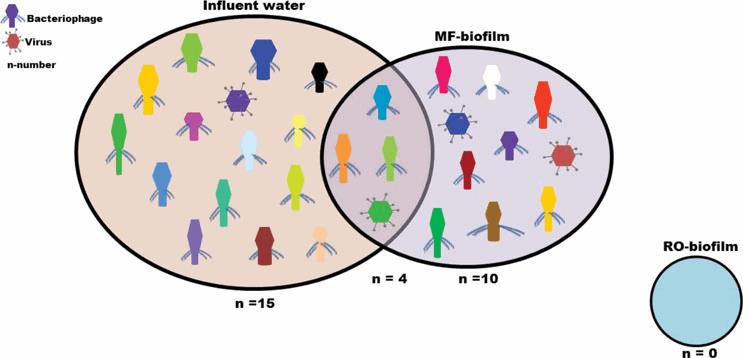
Figure 1. Venn diagram of total number of DNA viruses in secondary-treated wastewater that serves as the source water (influent) to GWRS, compared to MF and RO biofilm
For any of these approaches to achieve potable reuse, it is critical that water quality be accurately understood and monitored chemically and microbiologically. Regulatory standards for potable reuse vary by location. In California, for example, the regulations require 12-, 10-, and 10-log10 removal of viruses, Cryptosporidium, and Giardia, respectively, before potable reuse of recycled water. These water quality standards protect public health by ensuring that recycled water does not contain pathogenic microorganisms. This approach has been successfully used to ensure safety of conventional drinking water as well as recycled water for many decades. Looking to the future, the propagation of more advanced techniques for water quality characterization and monitoring — such as highthroughput sequencing — will provide a comprehensive and accurate assessment of microbial water quality, including potable reuse where microbial water quality is of particular concern.
Microbial Community Analysis
In nature, various microorganisms are found living together in complex mixtures in the same environment. These microbial communities can be highly variable in both composition and abundance of the constituent microbial species. Early studies to assess microbial communities used growth-dependent methods (Table 1). However, these methods identify only those organisms that can be grown in the laboratory when, in fact, the majority of microorganisms in the environment are difficult to grow (Staley and Konopka 1985; Rappe and Giovannoni 2003).
To detect known species of interest, such as pathogens, researchers and utilities often use polymerase chain reaction (PCR). In this method, microorganisms are collected from an environment (e.g., via filtration of water), the bacteria are broken open, and their DNA extracted. The segments of DNA matching to a specific sequence of interest are copied repeatedly using an enzyme called DNA polymerase. This amplification process allows researchers to detect DNA sequences of interest in a sample, even if they are at very low prevalence. PCR used in this manner requires specific, specially optimized probes and thus requires a priori knowledge of the DNA sequence of interest. This method can be used to detect anything for which the DNA sequence is known, including many bacterial, viral, or fungal pathogens, as well as antibiotic resistance genes (ARGs). A more advanced method, called quantitative PCR (qPCR), uses calibration to a known standard to determine the exact number of copies of DNA within the original sample. This number corresponds to the number of organisms in the sample. PCR and qPCR have the advantages of detecting and quantifying specific microorganisms, respectively, regardless of whether the organisms can be grown in the laboratory. However, for many targets, a separate reaction must be performed for each species of interest, and these methods can only be used where DNA sequences are already known (i.e., requires a priori knowledge).
In recent years, high-throughput DNA sequencing technologies (e.g., Illumina, Thermo Fisher, PacBio, and Oxford Nanopore), have been developed that allow sequencing all the DNA present in a sample. Through computational methods (bioinformatics), the large volume of data generated by sequencing can be used to identify and characterize all microbial species within a community. This process is independent of whether the microorganisms can be cultured in the laboratory. However, sequencing alone does not determine viability of the organisms, only that their DNA is present (in other words, those organisms could be alive or dead).
The most commonly employed sequencing approach used to identify and analyze microbial communities is 16S ribosomal RNA (rRNA) gene amplicon sequencing (see Table 1). This method usually focuses on bacteria and uses a “fingerprint” region of DNA that is unique among various bacteria but similar enough across all bacteria to be targeted by PCR. To perform amplicon sequencing, samples are collected and DNA is extracted. Using semispecific probes targeted to the fingerprint region of interest, the DNA is amplified using PCR. The amplified DNA fragments, known as amplicons, are then sequenced using high-throughput sequencing. Amplicon sequencing allows identification of bacteria that are present in very low numbers, as well as those that make up the majority of the microbial community. 16S rRNA gene amplicon sequencing for bacteria is widely used in surveys of microbial communities, including in the Human Microbiome Project (HMP, Human Microbiome Project Consortium 2012) and in the Earth Microbiome Project (http://www.earthmicrobiome.org/).
This method is rapid, relatively inexpensive, and, due to the amplification process, can be performed on samples with very little biomass, such as highly purified water. Using amplicon sequencing, bacteria can be classified to the level of genus; the sequence-based fingerprint isn’t specific enough to identify species or strain. Thus, pathogenic bacteria may not be distinguishable from their nonpathogenic relatives using this method. Depending on the study’s objectives, this can be an important limitation, notably for accurate detection and characterization of pathogens.
A more precise approach, called metagenomics (see Table 1), uses high-throughput sequencing to recover the whole genomes of all organisms present in a sample, rather than only a single fingerprint region. This type of sequencing captures data about all types of microorganisms, including bacteria, viruses, fungi, and protozoa, and can identify them to species, subspecies, and strain level. Additionally, metagenomics can provide information on antibiotic resistance, pathogenicity, and virulence factors associated with organisms in the sample. This method can also establish the presence of those organisms involved in nitrification, sulfide-related corrosion, and other processes related to microbial water quality. In one of its earliest applications, metagenomics was used to learn about the microbial causes of acid mine drainage (Tyson et al., 2004), and more recently it has helped researchers discover new bacteria in wastewater (Sekiguchi et al., 2015), as well as identify nitrifiers in drinking water (Pinto et al., 2016).
Table 1. Microbial Community Analysis Methods To Characterize Microorganisms In Water And Biofilms
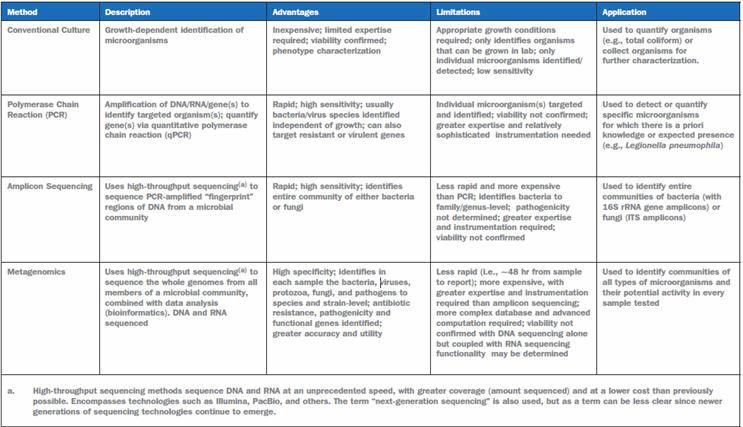
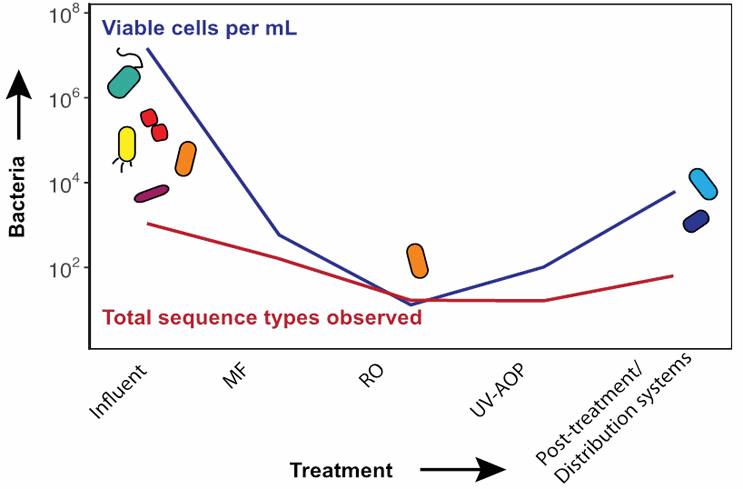
Figure 2. Changes in microbial water quality during advanced treatment. Influent is secondary-treated wastewater.
Compared to amplicon sequencing, metagenomics is more precise in the identification and abundance information it provides. Because of this, metagenomics is better suited for source-tracking investigations and assessing public health risks posed by microorganisms. However, metagenomics (in this early stage of its application) can also be more costly and time-consuming and usually requires more starting biomass, meaning larger volumes of water may be needed for analysis. The cost of metagenomics is decreasing relatively rapidly.
Both amplicon sequencing and metagenomics are used to identify microorganisms in microbial communities rapidly, accurately, and actionably. Selection of one approach over the other depends on the study’s objectives and resources available.
Whole Genome Studies Underway At OCWD’s Water Recycling Facility
Scientists at OCWD are using a metagenomic approach to identify and compare microbial communities in water reuse. In an initial proof-of-concept study, samples of secondary-treated wastewater (feedwater to the GWRS) were characterized, along with samples of biofilms from the feed side of the MF and RO membranes, using high-throughput sequencing to recover the whole genomes of all organisms present in the samples (Leddy et al., 2017). The investigation showed that certain viruses and bacteriophages (viruses that infect bacteria) were unique to the MF biofilm, compared to the feed (influent) water (Figure 1), while no viruses or bacteriophages were detected in the RO biofilm, among other findings. Subsequently, additional water samples were collected at each stage of treatment in the threestep GWRS process (MF, RO, and UV-AOP) for a study funded by the U.S. Bureau of Reclamation. That study aims to further characterize microbial communities in the reuse facility and document the degree of water quality improvement with each stage of treatment (i.e., reductions in microbial abundance and diversity, as well as removal of specific pathogens and reduction of microbial properties such as antibiotic resistance, etc.), with a final report expected in early 2018.
In a concurrent study, OCWD is using metagenomics to understand differences in microbial communities between GWRS and a more conventional drinking water treatment plant. Microbial communities of fungi, protozoa, bacteria, viruses, pathogens, and nonpathogens associated with treatment processes such as ozone and granular activated carbon (GAC) at the plant are being compared to the process used in GWRS. The source water reservoir to the conventional drinking water plant receives upstream treated wastewater discharges. At both facilities, many organisms that are common to wastewater have been identified, and the number and diversity of microbial species were found to substantially decrease after treatment. Metagenomics is being used to understand what microorganisms are associated with a particular treatment process, as well as their relative numbers in given samples and their potential activity and role.
Research On Treated Water Augmentation And The Distribution System
Researchers at the University of California, Berkeley, are also investigating the microorganisms found throughout advanced treatment trains for water reuse. Currently, they are evaluating two demonstration-scale potable reuse facilities in the U.S. using high-throughput sequencing methods, complemented by advanced techniques for quantifying live and dead microorganisms. They have found that bacterial concentrations decreased substantially through advanced treatment: Secondary wastewater (the source water) can contain as many as 10 million live cells per milliliter, while the final advanced treated water contains approximately 100 live cells per milliliter, or 0.001 percent of the initial concentration (Figure 2). This finding is important because bacterial quantity can impact overall microbial water quality. Using 16S rRNA gene amplicon sequencing, they found that the bacterial diversity, or the number of different types of bacteria present, decreases throughout advanced treatment. As many as 600 different types of bacteria may be present in secondary wastewater, while fewer than 65 types of bacteria (10 percent of the initial number) were found in purified water. Through their ongoing metagenomics efforts, researchers hope to learn more about why particular organisms are found in purified water.
Compared to conventional drinking water, purified water contains much lower levels of microorganisms and other nutrients (as was found in the metagenomics analyses for the OCWD study described above) but is still capable of supporting bacterial growth, as observed in model distribution systems (Figure 2). In some locations, such as a planned water reuse project in El Paso, TX, purified water will be introduced directly into existing drinking water distribution systems (i.e., treated water augmentation). Given that conventional drinking water and pipes in the distribution system contain their own microbial communities, the researchers are investigating what will happen when microbial communities from purified water and conventional drinking water mix together in the distribution system. Specifically, the team is investigating the effects that the new water might have on the occurrence of opportunistic pathogens, nitrification, and overall water quality in the distribution system. This work will help to identify potential benefits as well as unintended consequences of more direct potable reuse and recommend strategies for utilities adopting this new practice.
Conclusions
Microbial community analysis is rapidly advancing with the development and application of tools such as amplicon sequencing and metagenomics to identify and study microbial communities present in a given environment. These technologies provide an incredible amount of information that can be used to define the microbial signature of potable reuse treatment facilities and the subsequent distribution system both under normal operating conditions and to understand variabilities in performance.
References
- Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature, 486(7402), 207-214.
- Leddy, M. B., Hasan, N., Subramanian, P., Heberling, C., Cotruvo, J., and Colwell, R. R., 2017. Characterization of Microbial Signatures From Advanced Treated Wastewater Biofilms. Journal-American Water Works Association, 109(11), E503-E512.
- Pinto, A. J., Marcus, D. N., Ijaz, U. Z., Bautista-de lose Santos, Q. M., Dick, G. J., and Raskin, L. (2016). Metagenomic Evidence for the Presence of Comammox Nitrospira-Like Bacteria in a Drinking Water System. mSphere, 1(1). http://doi.org/10.1128/mSphere.00054-15
- Rappé, M.S., Giovannoni, S.J., 2003. The Uncultured Microbial Majority. Annual Review of Microbiology, 57, 369-394.
- Sekiguchi, Y., Ohashi, A., Parks, D. H., Yamauchi, T., Tyson, G. W., and Hugenholtz, P. (2015). First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ, 3, e740. http://doi.org/10.7717/peerj.740
- Staley, J.T., Konopka, A., 1985. Measurement of in Situ Activities of Nonphotosynthetic Microorganisms in Aquatic and Terrestrial Habitats. Annual Review of Microbiology, 39, 321-346. http://doi.org/10.1146/annurev.mi.39.100185.001541
- Tyson, G. W., Chapman, J., Hugenholtz, P., Allen, E. E., Ram, R. J., Richardson, P. M., et al. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature, 428(6978), 37–43. http://doi.org/10.1038/nature02340
About The Authors
Menu B. Leddy Principal Scientist, Research & Development Department, Orange County Water District, Fountain Valley, CA
Megan H. Plumlee Director of Research, Research & Development Department, Orange County Water District, Fountain Valley, CA
Rose S. Kantor Postdoctoral Fellow, Civil & Environmental Engineering Department, UC Berkeley, Berkeley, CA
Kara L. Nelson Professor, Civil & Environmental Engineering Department, UC Berkeley, Berkeley, CA
Scott E. Miller Graduate Student, Civil & Environmental Engineering Department, UC Berkeley, Berkeley, CA
Lauren C. Kennedy Graduate Student, Civil & Environmental Engineering Department, UC Berkeley, Berkeley, CA
Blake W. Stamps Postdoctoral Fellow, Civil & Environmental Engineering Department, Colorado School of Mines, Golden, CO
John R. Spear Professor, Civil & Environmental Engineering Department, Colorado School of Mines, Golden, CO
Nur A. Hasan Adjunct Associate Professor, Center of Bioinformatics and Computational Biology, University of Maryland, College Park, MD, and Vice President, CosmosID, Inc., Rockville, MD
Rita R. Colwell Distinguished University Professor, Center of Bioinformatics and Computational Biology, University of Maryland, College Park, MD; Johns Hopkins Bloomberg School of Public Health; and Founder, CosmosID Inc., Rockville, MD