
Emerging Best Practices For Contamination Management In Pharmaceutical Manufacturing
By Pavan Kumar Lakkakula, Beroe Inc.

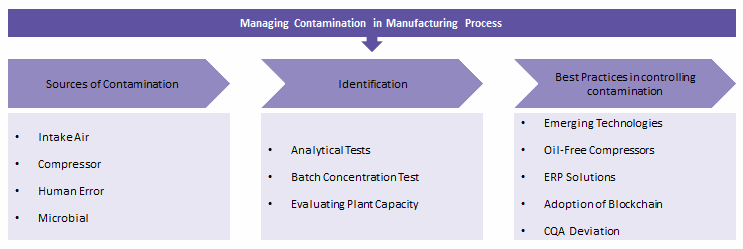
Despite quality audits and third-party oversight, contamination of prescription and over-the-counter (OTC) pharmaceutical products is still prevalent. This is despite the adoption of cGMP practices, SOPs, risk mitigation methods, and metrics such as key performance indicators. We evaluated various emerging practices to overcome contamination issues. Novel batch concentration tests and dedicated capacity utilization have proven to overcome the source of contaminants. For technology adoption in continuous manufacturing, which is unclear in terms of compliance, the FDA Emerging Technology Team could be used for advisory oversight. Other practices adopted include facility revisions on the use of oil-free air compressors, technologies such as detailed enterprise solutions, block chain, and measurement of critical attribute deviations. These have proven successful in both large and small molecule manufacturing processes. Retrospectively analysis to trace the source of contaminants, and therefore achieve increasingly contaminant-free finished goods, could be possible.
Identification Of The Source Of Contaminants
- Analytical Test Revisions: Packaging material that comes in direct contact with OTC products needs to have a leachability test performed as a randomized test. Water used for washing and sterilizing packaging or related materials needs to be contaminant-free.
- Batch Concentration Tests: Key contaminants with major adverse event effects are profiled, and relevant solvent is used to concentrate the contaminant from a sample taken from a larger batch. This allows the source of contamination to be detected when the contaminant is present at below-detectable levels. It is useful when multiple suppliers deliver products with below-detectable limits of contaminants.
- Dedicated Capacity Utilization: Plant audits check the number of chemicals manufactured per plant to evaluate if the plant manufactures multiple chemicals or if the chemical is from one of the multiple by-products.
Use Of The FDA Emerging Technology Team
The FDA Emerging Technology Team accepts proposals on the use of new technologies to be reviewed prior to regulatory submission. This team focuses on working with pharmaceutical quality audit teams toward implementation of new technologies. New technologies such as gene expression and protein production platforms and sensitive antibody drug conjugate platforms could be evaluated with the FDA during a continuous manufacturing program to answer late-stage questions that may arise during approvals. This also allows an external monitoring agency to ask the right questions in advance that would allow the manufacturer to implement the expectations in SOPs for manufacturing and audits.
Facility Contaminant Risk Overcome By Oil-Free Compressors
Although a lot of emphasis is placed on air filters, such as the material and pore size of the filter, little has been placed on the compressor systems for air quality. Atmospheric air and lubricant oils have received a lot of attention as being the source of contaminants. Oil-free compressors are the new trend to prevent contamination. Both partial and fully oil-free compressors are widely employed. Certain manufacturers can reduce overall lubricant usage as well in the compressors and save on operational oversight in managing standard oil utilization.
Detailed Enterprise Technology Solutions Could Trace Contaminants
Most companies have adopted ERP systems that work across multiple industry service lines. Adoption of pharma manufacturing ERP systems with detailed compliance oversight has been relatively less preferred than simpler, cost-efficient models. Looking at formulation management, operation management, and real-time information allows retrospective audits to identify the source of contaminants and make data-driven decisions. Pharma manufacturing-specific ERP solutions have specific features, such as FDA 21 CFR part 11 compliance sampling plans, stability testing, batch monitoring activities, electronic execution of SOPs, and QA as per cGMP standards. This has proven to support complete traceability, starting from the raw material to the finished product and packaging.
Ensure that your products are free of microbial contaminants by learning the simple steps to improve your aseptic processing techniques in the course:
Understanding Aseptic Technique and Cleanroom Behavior – Avoiding Human Error
Adoption Of Blockchain To Oversee OTC Batches
In certain cases, the source of the contaminant is a counterfeit drug or batches that have not been recorded in inventory. The use of RFID chips is costly. Rubix from Deloitte has noted the blockchain could impact drug safety, drug channels, and public safety. In addition, other startups such as iSolve, Blockverify, and Chronicled are focused on the pharmaceutical supply chain. Chronicled launched CryptoSeal, which are registered, adhesive seal strips containing a near-field communication (NFC) chip that can be customized per the manufacturer.
Overcoming Critical Quality Attributes (CQA) Deviation
CQA deviation from range could be due to several reasons, including aggregate content and degradation of raw material. The common factors reviewed and profiled are insoluble particulate matter impurities and the extent of contact between the single-use component and the batch including other endotoxins and microbial particulates. Risk evaluation in this case has been historically justified within permissible limits. However, if the frequency of risk were maintained as a stringent protocol, the chances of CQA deviation could be almost eliminated.
Conclusion
Exposure to contaminants during manufacturing can lead to cross-contamination, lower site ratings, and loss of revenue for manufacturers. It is therefore critical for companies to employ established and novel methods and procedures to prevent contamination. By ensuring that quality personnel are trained and audited in technology advances and novel processes, manufacturers can reduce contamination and minimize risk.
References:
- https://www.pharmasalmanac.com/articles/fda-finalizes-emerging-drug-manufacturing-technology-guidance
- http://www.oemupdate.com/technology/pumps-for-pharma/
- http://www.nasdaq.com/article/blockchain-technology-can-help-reduce-flow-of-counterfeit-drugs-cm721230
- Lovsin Barle, E., Bizec, J.C., Glogovac, M., Gromek, K., Winkler, G.C. “Determination and application of the permitted daily exposure (PDE) for topical ocular drugs in multipurpose manufacturing facilities”. Pharm Dev Technol. 2017 Apr 2:1doi:10.1080/10837450.2017.1312442. [Epub ahead of print] PubMed PMID: 28361586.
- Gordon, O., Goverde, M., Staerk, A., Roesti, D. “Validation of Milliflex(®) Quantum, for Bioburden Testing of Pharmaceutical Products”. PDA J Pharm Sci Technol. 2017 May-Jun;71(3):206-224. doi: 10.5731/pdajpst.2016.007450. Epub 2017 Feb 14. PubMed PMID: 28196917.
- Eissa, M.E. “Quantitative Microbial Risk Assessment of Pharmaceutical Products”. PDA J Pharm Sci Technol. 2017 May-Jun;71(3):245-251. doi: 10.5731/pdajpst.2016.007047. Epub 2016 Dec 14. PubMed PMID: 27974628.
- Lu, T.L., Pugach, O., Somerville, R., Rosenberg, S.A., Kochendefer, J.N., Better, M., Feldman, S.A. “A Rapid Cell Expansion Process for Production of Engineered Autologous CAR-T Cell Therapies”. Hum Gene Ther Methods. 2016 Dec;27(6):209-218. PubMed PMID: 27897048.
- Bercu, J.P., Morinello, E.J., Sehner, C., Shipp, B.K., Weideman, P.A. “Point of departure (PoD) selection for the derivation of acceptable daily exposures (ADEs) for active pharmaceutical ingredients (APIs).” Regul Toxicol Pharmacol. 2016 Aug;79 Suppl 1:S48-56. doi: 0.1016/j.yrtph.2016.05.028. Epub 2016 May 24. Review. PubMed PMID: 27233925.
- Araya, S., Lovsin-Barle, E., Glowienke, S. “Mutagenicity assessment strategy for pharmaceutical intermediates to aid limit setting for occupational exposure”. Regul Toxicol Pharmacol. 2015 Nov;73(2):515-20. doi: 10.1016/j.yrtph.2015.10.001. Epub 2015 Oct 8. PubMed PMID: 26454093.
- Ishii-Watabe, A., Hirose, A., Katori, N., Hashii, N., Arai, S., Awatsu, H., Eiza, A., Hara, Y., Hattori, H., Inoue, T., Isono, T., Iwakura, M., Kajihara, D., Kasahara, N., Matsuda, H., Murakami, S., Nakagawa, T., Okumura, T., Omasa, T., Takuma, S., Terashima, I., Tsukahara, M., Tsutsui, M., Yano, T., Kawasaki, N. “Approaches to Quality Risk Management When Using Single-Use Systems in the Manufacture of Biologics”. AAPS PharmSciTech. 2015 Oct;16(5):993-1001. doi: 10.1208/s12249-015-0368-z. Epub 2015 Aug 20. PubMed PMID: 26288941; PubMed Central PMCID: PMC4674654.
- ISO 14644-7 Cleanrooms and Associated Controlled Environments—Part 7: Separative Devices (Clean Air Hoods, Gloveboxes, Isolators and Mini Environments) 2004. [(accessed on 15 August 2016)]. Available online: https://www.iso.org/obp/ui/#iso:std:iso:14644:-7:ed-1:v1:en.
- ISO 10648-2 Containment Enclosures—Part 2: Classification According to Leak Tightness and Associated Checking Methods. [(accessed on 15 August 2016)]; Available online: https://www.iso.org/obp/ui/#iso:std:iso:10648:-2:ed-1:v1:en.
- ASTM E2930 Standard Practice for Pressure Decay Leak Test Method 2013. [(accessed on 15 August 2016)]. Available online: https://www.astm.org/Standards/E2930.htm.
- ISO 14644-3 Cleanrooms and Associated Controlled Environments—Part 3: Test Methods, B.4 Airflow Test 2005. [(accessed on 17 August 2016)].
About The Author:
 Pavan Kumar Lakkakula is a senior analyst of healthcare R&D for Beroe Inc. He specializes in the pharmaceutical category of product development services. Previously, he worked as an associate business intelligence analyst for QuintilesIMS. He has a master’s degree in pharmacy with a background in pharmaceutics from Tamil Nadu Dr. M.G.R. Medical University in Chennai, India. You can contact him at pavankumar.l@beroe-inc.com.
Pavan Kumar Lakkakula is a senior analyst of healthcare R&D for Beroe Inc. He specializes in the pharmaceutical category of product development services. Previously, he worked as an associate business intelligence analyst for QuintilesIMS. He has a master’s degree in pharmacy with a background in pharmaceutics from Tamil Nadu Dr. M.G.R. Medical University in Chennai, India. You can contact him at pavankumar.l@beroe-inc.com.